Knowledge-primed Neural Networks (KPNNs) for single cell data#
In this tutorial we will show how CORNETO can be used to build custom neural network architectures informed by prior knowledge. We will see how to implement a knowledge-primed neural network1. We will use the single cell data from the publication “Knowledge-primed neural networks enable biologically interpretable deep learning on single-cell sequencing data”, from Nikolaus Fortelny & Christoph Bock, where they used a single-cell RNA-seq dataset they previously generated2, which measures cellular responses to T cell receptor (TCR) stimulation in a standardized in vitro model. The dataset was chosen due to the TCR signaling pathway’s complexity and its well-characterized role in orchestrating transcriptional responses to antigen detection in T cells.
Why CORNETO?#
In the original publication, authors built a KPNN by searching on databases, building a Direct Acyclic Graph (DAG) by running shortest paths from TCR receptor to genes. However, this approach is not optimal. CORNETO, thanks to its advanced capabilities for modeling and optimization on networks, provides methods to automatically find DAG architectures in an optimal way.
In addition to this, CORNETO provides methods to build DAG NN architectures with ease using Keras +3, making KPNN implementation very flexible and interoperable with backends like Pytorch, Tensorflow and JAX.
How does it work?#
Thanks to CORNETO’s building blocks for optimization over networks, we can easily model optimization problems to find DAG architectures from a Prior Knowledge Network. After we have the backbone, we can convert it to a neural network using the utility functions included in CORNETO.
References#
Fortelny, N., & Bock, C. (2020). Knowledge-primed neural networks enable biologically interpretable deep learning on single-cell sequencing data. Genome biology, 21, 1-36.
Datlinger, P., Rendeiro, A. F., Schmidl, C., Krausgruber, T., Traxler, P., Klughammer, J., … & Bock, C. (2017). Pooled CRISPR screening with single-cell transcriptome readout. Nature methods, 14(3), 297-301.
Download and import the single cell dataset#
import os
import tempfile
import urllib.parse
import urllib.request
import numpy as np
import pandas as pd
import scanpy as sc
import corneto as cn
with urllib.request.urlopen("http://kpnn.computational-epigenetics.org/") as response:
web_input = response.geturl()
print("Effective URL:", web_input)
files = ["TCR_Edgelist.csv", "TCR_ClassLabels.csv", "TCR_Data.h5"]
temp_dir = tempfile.mkdtemp()
# Download files
file_paths = []
for file in files:
url = urllib.parse.urljoin(web_input, file)
output_path = os.path.join(temp_dir, file)
print(f"Downloading {url} to {output_path}")
try:
with urllib.request.urlopen(url) as response:
with open(output_path, "wb") as f:
f.write(response.read())
file_paths.append(output_path)
except Exception as e:
print(f"Failed to download {url}: {e}")
print("Downloaded files:")
for path in file_paths:
print(path)
Effective URL: https://medical-epigenomics.org/papers/fortelny2019/
Downloading https://medical-epigenomics.org/papers/fortelny2019/TCR_Edgelist.csv to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp5w2c_81g/TCR_Edgelist.csv
Downloading https://medical-epigenomics.org/papers/fortelny2019/TCR_ClassLabels.csv to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp5w2c_81g/TCR_ClassLabels.csv
Downloading https://medical-epigenomics.org/papers/fortelny2019/TCR_Data.h5 to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp5w2c_81g/TCR_Data.h5
Downloaded files:
/var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp5w2c_81g/TCR_Edgelist.csv
/var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp5w2c_81g/TCR_ClassLabels.csv
/var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp5w2c_81g/TCR_Data.h5
# The data contains also the original network they built with shortest paths.
# We will use it to replicate the study
df_edges = pd.read_csv(file_paths[0])
df_labels = pd.read_csv(file_paths[1])
# Import the 10x data with Scanpy
adata = sc.read_10x_h5(file_paths[2])
df_labels
| barcode | TCR | |
|---|---|---|
| 0 | AAACCTGCACACATGT-1 | 0 |
| 1 | AAACCTGCACGTCTCT-1 | 0 |
| 2 | AAACCTGTCAATACCG-1 | 0 |
| 3 | AAACCTGTCGTGGTCG-1 | 0 |
| 4 | AAACGGGTCTGAGTGT-1 | 0 |
| ... | ... | ... |
| 1730 | TTTCCTCGTCATGCCG-2 | 1 |
| 1731 | TTTGCGCGTAGCCTCG-2 | 1 |
| 1732 | TTTGGTTAGATACACA-2 | 1 |
| 1733 | TTTGGTTGTATGAATG-2 | 1 |
| 1734 | TTTGGTTTCCAAGTAC-2 | 1 |
1735 rows × 2 columns
df_edges
| parent | child | |
|---|---|---|
| 0 | TCR | ZAP70 |
| 1 | ZAP70 | MAPK14 |
| 2 | MAPK14 | FOXO3 |
| 3 | MAPK14 | STAT1 |
| 4 | MAPK14 | STAT3 |
| ... | ... | ... |
| 27574 | HMGA1 | MTRNR2L9_gene |
| 27575 | MYB | C12orf50_gene |
| 27576 | MYB | TRPC5OS_gene |
| 27577 | SOX2 | TRPC5OS_gene |
| 27578 | CRTC1 | MTRNR2L9_gene |
27579 rows × 2 columns
adata.var
| gene_ids | |
|---|---|
| DDX11L1 | ENSG00000223972 |
| WASH7P | ENSG00000227232 |
| MIR6859-2 | ENSG00000278267 |
| MIR1302-10 | ENSG00000243485 |
| MIR1302-11 | ENSG00000274890 |
| ... | ... |
| Tcrlibrary_RUNX2_3_gene | Tcrlibrary_RUNX2_3_gene |
| Tcrlibrary_ZAP70_1_gene | Tcrlibrary_ZAP70_1_gene |
| Tcrlibrary_ZAP70_2_gene | Tcrlibrary_ZAP70_2_gene |
| Tcrlibrary_ZAP70_3_gene | Tcrlibrary_ZAP70_3_gene |
| Cas9_blast_gene | Cas9_blast_gene |
64370 rows × 1 columns
# We can normalize the data, however, it is better to avoid
# preprocessing the whole dataset before splitting in training and test
# to avoid data leakage.
# NOTE: Normalization can be done inside the cross-val loop
# sc.pp.normalize_total(adata, target_sum=1e6)
# Log-transform the data does not leak data as it does not estimate anything
sc.pp.log1p(adata)
adata.obs
| AAACCTGAGAAACCAT-1 |
|---|
| AAACCTGAGAAACCGC-1 |
| AAACCTGAGAAACCTA-1 |
| AAACCTGAGAAACGAG-1 |
| AAACCTGAGAAACGCC-1 |
| ... |
| TTTGTCATCTTTACAC-2 |
| TTTGTCATCTTTACGT-2 |
| TTTGTCATCTTTAGGG-2 |
| TTTGTCATCTTTAGTC-2 |
| TTTGTCATCTTTCCTC-2 |
1474560 rows × 0 columns
barcodes = adata.obs_names
barcodes
Index(['AAACCTGAGAAACCAT-1', 'AAACCTGAGAAACCGC-1', 'AAACCTGAGAAACCTA-1',
'AAACCTGAGAAACGAG-1', 'AAACCTGAGAAACGCC-1', 'AAACCTGAGAAAGTGG-1',
'AAACCTGAGAACAACT-1', 'AAACCTGAGAACAATC-1', 'AAACCTGAGAACTCGG-1',
'AAACCTGAGAACTGTA-1',
...
'TTTGTCATCTTGGGTA-2', 'TTTGTCATCTTGTACT-2', 'TTTGTCATCTTGTATC-2',
'TTTGTCATCTTGTCAT-2', 'TTTGTCATCTTGTTTG-2', 'TTTGTCATCTTTACAC-2',
'TTTGTCATCTTTACGT-2', 'TTTGTCATCTTTAGGG-2', 'TTTGTCATCTTTAGTC-2',
'TTTGTCATCTTTCCTC-2'],
dtype='object', length=1474560)
gene_names = adata.var.index
print(gene_names)
Index(['DDX11L1', 'WASH7P', 'MIR6859-2', 'MIR1302-10', 'MIR1302-11', 'FAM138A',
'OR4G4P', 'OR4G11P', 'OR4F5', 'RP11-34P13.7',
...
'Tcrlibrary_RUNX1_1_gene', 'Tcrlibrary_RUNX1_2_gene',
'Tcrlibrary_RUNX1_3_gene', 'Tcrlibrary_RUNX2_1_gene',
'Tcrlibrary_RUNX2_2_gene', 'Tcrlibrary_RUNX2_3_gene',
'Tcrlibrary_ZAP70_1_gene', 'Tcrlibrary_ZAP70_2_gene',
'Tcrlibrary_ZAP70_3_gene', 'Cas9_blast_gene'],
dtype='object', length=64370)
len(set(df_labels.barcode.tolist()))
1735
len(set(barcodes.tolist()))
1474560
matched_barcodes = sorted(set(barcodes.tolist()) & set(df_labels.barcode.tolist()))
len(matched_barcodes)
1735
# This is the InPathsY data in the original code of KPNNs
df_labels
| barcode | TCR | |
|---|---|---|
| 0 | AAACCTGCACACATGT-1 | 0 |
| 1 | AAACCTGCACGTCTCT-1 | 0 |
| 2 | AAACCTGTCAATACCG-1 | 0 |
| 3 | AAACCTGTCGTGGTCG-1 | 0 |
| 4 | AAACGGGTCTGAGTGT-1 | 0 |
| ... | ... | ... |
| 1730 | TTTCCTCGTCATGCCG-2 | 1 |
| 1731 | TTTGCGCGTAGCCTCG-2 | 1 |
| 1732 | TTTGGTTAGATACACA-2 | 1 |
| 1733 | TTTGGTTGTATGAATG-2 | 1 |
| 1734 | TTTGGTTTCCAAGTAC-2 | 1 |
1735 rows × 2 columns
Import PKN with CORNETO#
cn.info()

|
|
outputs_pkn = list(set(df_edges.parent.tolist()) - set(df_edges.child.tolist()))
inputs_pkn = set(df_edges.child.tolist()) - set(df_edges.parent.tolist())
input_pkn_genes = list(set(g.split("_")[0] for g in inputs_pkn))
len(inputs_pkn), len(outputs_pkn)
(13121, 1)
tuples = [(r.child, 1, r.parent) for _, r in df_edges.iterrows()]
G = cn.Graph.from_sif_tuples(tuples)
G = G.prune(inputs_pkn, outputs_pkn)
# Size of the original PKN provided by the authors
G.shape
(13439, 27579)
Select the single cell data for training#
adata_matched = adata[adata.obs_names.isin(matched_barcodes), adata.var_names.isin(input_pkn_genes)]
adata_matched.shape
(1735, 14229)
non_zero_genes = set(adata_matched.to_df().columns[adata_matched.to_df().sum(axis=0) >= 1e-6].values)
len(non_zero_genes)
12459
len(non_zero_genes.intersection(adata_matched.var_names))
12459
adata_matched = adata_matched[:, adata_matched.var_names.isin(non_zero_genes)]
# Many duplicates still 0 counts
adata_matched = adata_matched[:, adata_matched.to_df().sum(axis=0) != 0]
adata_matched.shape
(1735, 12487)
df_expr = adata_matched.to_df()
df_expr = df_expr.groupby(df_expr.columns, axis=1).max()
df_expr
| A1BG | A2ML1 | AAAS | AACS | AADAT | AAED1 | AAGAB | AAK1 | AAMDC | AAMP | ... | ZSWIM8 | ZUFSP | ZW10 | ZWILCH | ZXDC | ZYG11A | ZYG11B | ZYX | ZZEF1 | ZZZ3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AAACCTGCACACATGT-1 | 0.0 | 0.0 | 0.000000 | 0.693147 | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 1.098612 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| AAACCTGCACGTCTCT-1 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.693147 | 0.000000 | 0.000000 | 0.0 | 0.693147 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| AAACCTGTCAATACCG-1 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.693147 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| AAACCTGTCGTGGTCG-1 | 0.0 | 0.0 | 1.098612 | 0.000000 | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 1.098612 | ... | 0.000000 | 0.693147 | 0.000000 | 0.693147 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| AAACGGGTCTGAGTGT-1 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.693147 | 0.000000 | 0.000000 | 0.0 | 0.000000 | ... | 0.693147 | 0.000000 | 0.693147 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| TTTCCTCGTCATGCCG-2 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.000000 | 0.000000 | 0.693147 | 0.0 | 0.693147 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| TTTGCGCGTAGCCTCG-2 | 0.0 | 0.0 | 1.098612 | 0.000000 | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 1.386294 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| TTTGGTTAGATACACA-2 | 0.0 | 0.0 | 1.098612 | 0.000000 | 0.0 | 0.000000 | 0.000000 | 1.098612 | 0.0 | 0.693147 | ... | 0.693147 | 0.000000 | 0.693147 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.693147 | 0.693147 | 0.000000 |
| TTTGGTTGTATGAATG-2 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.693147 | 1.098612 | 0.000000 | 0.0 | 0.693147 | ... | 0.000000 | 0.693147 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.693147 |
| TTTGGTTTCCAAGTAC-2 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.693147 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 1.098612 | 0.000000 | 0.000000 |
1735 rows × 12459 columns
Building and training the KPNN#
Now we will use the provided PKN by the authors and the utility functions in CORNETO to build a KPNN similar to the one used in the original manuscript
import os
os.environ["KERAS_BACKEND"] = "jax"
import keras
from keras.callbacks import EarlyStopping
from sklearn.metrics import (
accuracy_score,
f1_score,
precision_score,
recall_score,
roc_auc_score,
)
from sklearn.model_selection import StratifiedKFold
# Use the data from the experiment
X = df_expr.values
y = df_labels.set_index("barcode").loc[df_expr.index, "TCR"].values
X.shape, y.shape
((1735, 12459), (1735,))
# We can prefilter on top N genes to make this faster
top_n = None
# From the given PKN
outputs_pkn = list(set(df_edges.parent.tolist()) - set(df_edges.child.tolist()))
inputs_pkn = set(df_edges.child.tolist()) - set(df_edges.parent.tolist())
input_pkn_genes = list(set(g.split("_")[0] for g in inputs_pkn))
if top_n is not None and top_n > 0:
input_pkn_genes = list(
set(input_pkn_genes).intersection(df_expr.var(axis=0).sort_values(ascending=False).head(top_n).index)
)
inputs_pkn = list(g + "_gene" for g in input_pkn_genes)
len(inputs_pkn), len(outputs_pkn)
(13121, 1)
input_nn_genes = list(set(input_pkn_genes).intersection(df_expr.columns))
input_nn = [g + "_gene" for g in input_nn_genes]
len(input_nn)
12459
# Build corneto graph
tuples = [(r.child, 1, r.parent) for _, r in df_edges.iterrows()]
G = cn.Graph.from_sif_tuples(tuples)
G = G.prune(input_nn, outputs_pkn)
G.shape
(12767, 25928)
len(input_nn), len(input_nn_genes)
(12459, 12459)
len(set(input_nn).intersection(G.V))
12459
X = df_expr.loc[:, input_nn_genes].values
y = df_labels.set_index("barcode").loc[df_expr.index, "TCR"].values
X.shape, y.shape
((1735, 12459), (1735,))
from corneto._ml import build_dagnn
def stratified_kfold(
G,
inputs,
outputs,
n_splits=5,
shuffle=True,
random_state=42,
lr=0.001,
patience=10,
file_weights="weights",
dagnn_config=dict(
batch_norm_input=True,
batch_norm_center=False,
batch_norm_scale=False,
bias_reg_l1=1e-3,
bias_reg_l2=1e-2,
dropout=0.20,
default_hidden_activation="sigmoid",
default_output_activation="sigmoid",
verbose=False,
),
):
kfold = StratifiedKFold(n_splits=n_splits, shuffle=shuffle, random_state=random_state)
models = []
metrics = {m: [] for m in ["accuracy", "precision", "recall", "f1", "roc_auc"]}
for i, (train_idx, val_idx) in enumerate(kfold.split(X, y)):
X_train, X_val = X[train_idx], X[val_idx]
y_train, y_val = y[train_idx], y[val_idx]
print("Building DAG NN model with CORNETO using Keras with JAX...")
print(f" > N. inputs: {len(input_nn)}")
print(f" > N. outputs: {len(outputs_pkn)}")
model = build_dagnn(G, input_nn, outputs_pkn, **dagnn_config)
print(f" > N. parameters: {model.count_params()}")
# Train the model with Adam
opt = keras.optimizers.Adam(learning_rate=lr)
early_stopping = EarlyStopping(monitor="val_loss", patience=patience, restore_best_weights=True)
print("Compiling...")
model.compile(optimizer=opt, loss="binary_crossentropy", metrics=["accuracy"])
print("Fitting...")
model.fit(
X_train,
y_train,
validation_data=(X_val, y_val),
epochs=200,
batch_size=64,
verbose=0,
callbacks=[early_stopping],
)
if file_weights is not None:
filename = f"{file_weights}_{i}.keras"
model.save(filename)
print(f"Weights saved to {filename}")
# Predictions and metrics calculation
y_pred_proba = model.predict(X_val).flatten()
y_pred = (y_pred_proba > 0.5).astype(int)
acc = accuracy_score(y_val, y_pred)
precision = precision_score(y_val, y_pred)
recall = recall_score(y_val, y_pred)
f1 = f1_score(y_val, y_pred)
roc_auc = roc_auc_score(y_val, y_pred_proba)
metrics["accuracy"].append(acc)
metrics["precision"].append(precision)
metrics["recall"].append(recall)
metrics["f1"].append(f1)
metrics["roc_auc"].append(roc_auc)
print(f" > Fold {i} validation ROC-AUC={roc_auc:.3f}")
models.append(model)
return models, metrics
temp_weights = tempfile.mkdtemp()
models, metrics = stratified_kfold(G, input_nn, outputs_pkn, file_weights=os.path.join(temp_weights, "weights"))
print("Validation metrics:")
for k, v in metrics.items():
print(f" - {k}: {np.mean(v):.3f}")
Building DAG NN model with CORNETO using Keras with JAX...
> N. inputs: 12459
> N. outputs: 1
> N. parameters: 26236
Compiling...
Fitting...
Weights saved to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp044vf9e8/weights_0.keras
1/11 ━━━━━━━━━━━━━━━━━━━━ 29s 3s/step
2/11 ━━━━━━━━━━━━━━━━━━━━ 17s 2s/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 0s 412ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 7s 412ms/step
> Fold 0 validation ROC-AUC=0.991
Building DAG NN model with CORNETO using Keras with JAX...
> N. inputs: 12459
> N. outputs: 1
> N. parameters: 26236
Compiling...
Fitting...
Weights saved to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp044vf9e8/weights_1.keras
1/11 ━━━━━━━━━━━━━━━━━━━━ 16s 2s/step
2/11 ━━━━━━━━━━━━━━━━━━━━ 12s 1s/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 0s 308ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 5s 308ms/step
> Fold 1 validation ROC-AUC=0.979
Building DAG NN model with CORNETO using Keras with JAX...
> N. inputs: 12459
> N. outputs: 1
> N. parameters: 26236
Compiling...
Fitting...
Weights saved to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp044vf9e8/weights_2.keras
1/11 ━━━━━━━━━━━━━━━━━━━━ 11s 1s/step
2/11 ━━━━━━━━━━━━━━━━━━━━ 8s 942ms/step
10/11 ━━━━━━━━━━━━━━━━━━━━ 0s 123ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 0s 208ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 3s 208ms/step
> Fold 2 validation ROC-AUC=0.982
Building DAG NN model with CORNETO using Keras with JAX...
> N. inputs: 12459
> N. outputs: 1
> N. parameters: 26236
Compiling...
Fitting...
Weights saved to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp044vf9e8/weights_3.keras
1/11 ━━━━━━━━━━━━━━━━━━━━ 11s 1s/step
2/11 ━━━━━━━━━━━━━━━━━━━━ 7s 886ms/step
10/11 ━━━━━━━━━━━━━━━━━━━━ 0s 116ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 0s 202ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 3s 202ms/step
> Fold 3 validation ROC-AUC=0.994
Building DAG NN model with CORNETO using Keras with JAX...
> N. inputs: 12459
> N. outputs: 1
> N. parameters: 26236
Compiling...
Fitting...
Weights saved to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp044vf9e8/weights_4.keras
1/11 ━━━━━━━━━━━━━━━━━━━━ 11s 1s/step
2/11 ━━━━━━━━━━━━━━━━━━━━ 9s 1s/step
10/11 ━━━━━━━━━━━━━━━━━━━━ 0s 132ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 0s 220ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 3s 220ms/step
> Fold 4 validation ROC-AUC=0.992
Validation metrics:
- accuracy: 0.964
- precision: 0.969
- recall: 0.958
- f1: 0.963
- roc_auc: 0.988
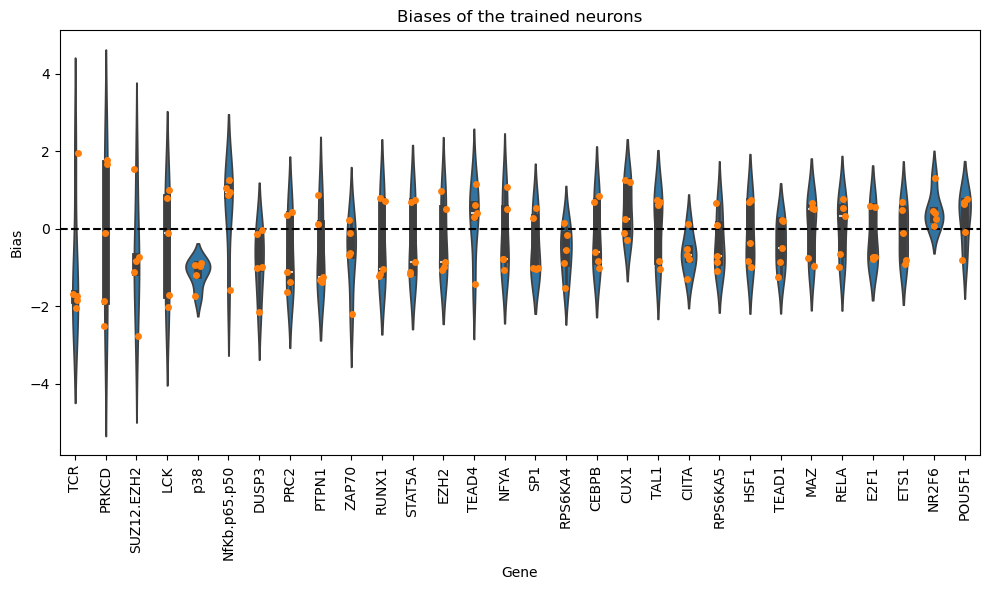
Now, we will analyze the learned biases for each of the nodes in the graph. Note that authors in the KPNN paper explain a way to extract weights for the nodes, based on the learned interactions and accounting for biases in the structure of the NN. Here we just show the learned biases of the nodes of the NN across 5 folds. Please be careful interpreting these weights.
# We collect the weights obtained in each fold
def load_biases(file="weights", folds=5):
biases = []
mean_inputs = []
for i in range(5):
model = keras.models.load_model(f"{file}_{i}.keras")
for layer in model.layers:
weights = layer.get_weights()
if weights:
biases.append((i, layer.name, weights[1][0]))
mean_inputs.append((i, layer.name, weights[0].mean()))
df_biases = pd.DataFrame(biases, columns=["fold", "gene", "bias"])
df_biases["abs_bias"] = df_biases.bias.abs()
df_biases["pow2_bias"] = df_biases.bias.pow(2)
df_biases = df_biases.set_index(["fold", "gene"])
return df_biases
df_biases = load_biases(file=os.path.join(temp_weights, "weights"), folds=5)
df_biases.sort_values(by="abs_bias", ascending=False).head(10)
| bias | abs_bias | pow2_bias | ||
|---|---|---|---|---|
| fold | gene | |||
| 3 | SUZ12.EZH2 | -2.768535 | 2.768535 | 7.664784 |
| 4 | PRKCD | -2.499154 | 2.499154 | 6.245770 |
| 1 | ZAP70 | -2.205527 | 2.205527 | 4.864350 |
| 3 | DUSP3 | -2.144189 | 2.144189 | 4.597546 |
| TCR | -2.033000 | 2.033000 | 4.133087 | |
| 4 | LCK | -2.014710 | 2.014710 | 4.059057 |
| TCR | 1.960717 | 1.960717 | 3.844413 | |
| 3 | PRKCD | -1.850238 | 1.850238 | 3.423379 |
| 0 | TCR | -1.836677 | 1.836677 | 3.373384 |
| PRKCD | 1.777246 | 1.777246 | 3.158603 |
df_biases_full = df_biases.copy().reset_index()
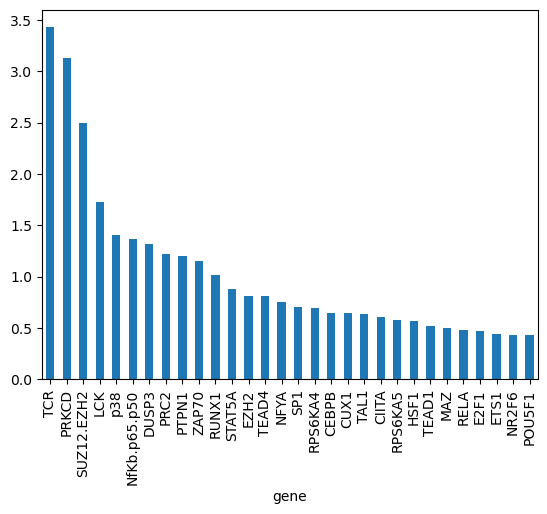
gene_biases_score = df_biases_full.groupby("gene")["pow2_bias"].mean().sort_values(ascending=False)
gene_biases_score
gene
TCR 3.427786
PRKCD 3.129591
SUZ12.EZH2 2.498496
LCK 1.725973
p38 1.404157
...
MLL2.complex 0.000493
HMT 0.000109
HSF2 0.000087
GATA3 0.000080
REST 0.000022
Name: pow2_bias, Length: 308, dtype: float32
gene_biases_score.head(30).plot.bar()
<Axes: xlabel='gene'>
import matplotlib.pyplot as plt
import pandas as pd
import seaborn as sns
# Get the top genes sorted by mean bias
top_genes = gene_biases_score.head(30).index
# Filter the DataFrame to include only rows with these top genes
filtered_df = df_biases_full[df_biases_full["gene"].isin(top_genes)]
plt.figure(figsize=(10, 6))
sns.violinplot(data=filtered_df, x="gene", y="bias", order=top_genes, dodge=True)
sns.stripplot(data=filtered_df, x="gene", y="bias", order=top_genes, dodge=True)
plt.xticks(rotation=90)
plt.title("Biases of the trained neurons")
plt.xlabel("Gene")
plt.ylabel("Bias")
plt.axhline(0, linestyle="--", color="k")
plt.tight_layout()
Use CORNETO for NN pruning#
Now we will show how CORNETO can be used to extract a smaller, yet complete DAG from the original PKN provided by the authors. We will add input edges to each input node and an output edge through TCR to indicate which nodes are the inputs and which one the output. We will use then Acyclic Flow to find the smallest DAG comprising these nodes
G_dag = G.copy()
new_edges = []
for g in input_nn:
new_edges.append(G_dag.add_edge((), g))
new_edges.append(G_dag.add_edge("TCR", ()))
print(G_dag.shape)
# Find small DAG. We use Acyclic Flow to find over the space of DAGs
P = cn.opt.AcyclicFlow(G_dag)
# We enforce that the input genes and the output gene are part of the solution
P += P.expr.with_flow[new_edges] == 1
# Minimize the number of active edges
P.add_objectives(sum(P.expr.with_flow), weights=1)
P.solve(solver="SCIP", verbosity=1, max_seconds=300);
(12767, 38388)
===============================================================================
CVXPY
v1.6.6
===============================================================================
-------------------------------------------------------------------------------
Compilation
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
Numerical solver
-------------------------------------------------------------------------------
Running HiGHS 1.8.0 (git hash: 222cce7): Copyright (c) 2024 HiGHS under MIT licence terms
Coefficient ranges:
Matrix [1e-04, 1e+04]
Cost [1e+00, 1e+00]
Bound [1e+00, 1e+00]
RHS [1e+00, 1e+04]
Presolving model
83375 rows, 57447 cols, 205995 nonzeros 0s
40079 rows, 41245 cols, 128297 nonzeros 0s
39352 rows, 38165 cols, 131120 nonzeros 0s
Objective function is integral with scale 1
Solving MIP model with:
39352 rows
38165 cols (18825 binary, 0 integer, 0 implied int., 19340 continuous)
131120 nonzeros
MIP-Timing: 0.54 - starting analytic centre calculation
Src: B => Branching; C => Central rounding; F => Feasibility pump; H => Heuristic; L => Sub-MIP;
P => Empty MIP; R => Randomized rounding; S => Solve LP; T => Evaluate node; U => Unbounded;
z => Trivial zero; l => Trivial lower; u => Trivial upper; p => Trivial point
Nodes | B&B Tree | Objective Bounds | Dynamic Constraints | Work
Src Proc. InQueue | Leaves Expl. | BestBound BestSol Gap | Cuts InLp Confl. | LpIters Time
0 0 0 0.00% 19693 inf inf 0 0 0 0 0.5s
0 0 0 0.00% 19693 inf inf 0 0 1 19806 1.8s
0 0 0 0.00% 22669.402362 inf inf 5152 3535 132 39839 7.3s
L 0 0 0 0.00% 22669.405102 25088 9.64% 5152 3535 258 39839 19.3s
0.2% inactive integer columns, restarting
Model after restart has 39282 rows, 38128 cols (18793 bin., 0 int., 0 impl., 19335 cont.), and 130976 nonzeros
0 0 0 0.00% 22669.405102 25088 9.64% 3297 0 0 92375 19.6s
0 0 0 0.00% 22669.405102 25088 9.64% 3297 3175 4 136853 33.4s
0 0 0 0.00% 22995.391228 25088 8.34% 4679 3772 4 146501 38.4s
Symmetry detection completed in 3.9s
Found 2264 generator(s)
55 0 1 0.00% 22996.391797 25088 8.34% 6285 3695 172 303808 149.4s
220 217 2 0.00% 23067.071606 25088 8.06% 6286 3695 481 316586 205.1s
250 216 3 0.00% 23067.071606 25088 8.06% 6394 3737 528 407997 278.4s
280 274 4 0.00% 23067.071606 25088 8.06% 6395 3737 613 413452 300.0s
280 274 4 0.00% 23067.071606 25088 8.06% 6395 3737 613 413452 300.0s
Solving report
Status Time limit reached
Primal bound 25088
Dual bound 23068
Gap 8.05% (tolerance: 0.01%)
P-D integral 23.3781609893
Solution status feasible
25088 (objective)
0 (bound viol.)
0 (int. viol.)
0 (row viol.)
Timing 300.04 (total)
0.00 (presolve)
0.00 (solve)
0.00 (postsolve)
Max sub-MIP depth 8
Nodes 280
Repair LPs 0 (0 feasible; 0 iterations)
LP iterations 413452 (total)
176391 (strong br.)
35258 (separation)
52536 (heuristics)
Solver terminated with message: Time limit reached. (HiGHS Status 13: Time limit reached)
-------------------------------------------------------------------------------
Summary
-------------------------------------------------------------------------------
G_subdag = G_dag.edge_subgraph(P.expr.with_flow.value > 0.5)
G_dag.shape, G_subdag.shape
((12767, 38388), (12629, 25088))
rel_dag_compression = (1 - (G_subdag.num_edges / G_dag.num_edges)) * 100
print(f"KPNN edge compression (0-100%): {rel_dag_compression:.2f}%")
KPNN edge compression (0-100%): 34.65%
pruned_models, pruned_metrics = stratified_kfold(
G_subdag,
input_nn,
outputs_pkn,
file_weights=os.path.join(temp_weights, "pruned_weights"),
)
print("Validation metrics:")
for k, v in pruned_metrics.items():
print(f" - {k}: {np.mean(v):.3f}")
Building DAG NN model with CORNETO using Keras with JAX...
> N. inputs: 12459
> N. outputs: 1
> N. parameters: 12798
Compiling...
Fitting...
Weights saved to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp044vf9e8/pruned_weights_0.keras
1/11 ━━━━━━━━━━━━━━━━━━━━ 5s 583ms/step
2/11 ━━━━━━━━━━━━━━━━━━━━ 4s 517ms/step
10/11 ━━━━━━━━━━━━━━━━━━━━ 0s 68ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 0s 111ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 2s 111ms/step
> Fold 0 validation ROC-AUC=0.994
Building DAG NN model with CORNETO using Keras with JAX...
> N. inputs: 12459
> N. outputs: 1
> N. parameters: 12798
Compiling...
Fitting...
Weights saved to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp044vf9e8/pruned_weights_1.keras
1/11 ━━━━━━━━━━━━━━━━━━━━ 6s 666ms/step
2/11 ━━━━━━━━━━━━━━━━━━━━ 4s 476ms/step
10/11 ━━━━━━━━━━━━━━━━━━━━ 0s 64ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 0s 108ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 2s 108ms/step
> Fold 1 validation ROC-AUC=0.977
Building DAG NN model with CORNETO using Keras with JAX...
> N. inputs: 12459
> N. outputs: 1
> N. parameters: 12798
Compiling...
Fitting...
Weights saved to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp044vf9e8/pruned_weights_2.keras
1/11 ━━━━━━━━━━━━━━━━━━━━ 6s 649ms/step
2/11 ━━━━━━━━━━━━━━━━━━━━ 5s 556ms/step
10/11 ━━━━━━━━━━━━━━━━━━━━ 0s 72ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 0s 121ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 2s 121ms/step
> Fold 2 validation ROC-AUC=0.986
Building DAG NN model with CORNETO using Keras with JAX...
> N. inputs: 12459
> N. outputs: 1
> N. parameters: 12798
Compiling...
Fitting...
Weights saved to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp044vf9e8/pruned_weights_3.keras
1/11 ━━━━━━━━━━━━━━━━━━━━ 6s 622ms/step
2/11 ━━━━━━━━━━━━━━━━━━━━ 4s 480ms/step
10/11 ━━━━━━━━━━━━━━━━━━━━ 0s 63ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 0s 106ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 2s 106ms/step
> Fold 3 validation ROC-AUC=0.995
Building DAG NN model with CORNETO using Keras with JAX...
> N. inputs: 12459
> N. outputs: 1
> N. parameters: 12798
Compiling...
Fitting...
Weights saved to /var/folders/b4/gwkwsdb93sv11rtztqbm3l040000gn/T/tmp044vf9e8/pruned_weights_4.keras
1/11 ━━━━━━━━━━━━━━━━━━━━ 5s 566ms/step
2/11 ━━━━━━━━━━━━━━━━━━━━ 4s 489ms/step
10/11 ━━━━━━━━━━━━━━━━━━━━ 0s 64ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 0s 107ms/step
11/11 ━━━━━━━━━━━━━━━━━━━━ 2s 107ms/step
> Fold 4 validation ROC-AUC=0.995
Validation metrics:
- accuracy: 0.964
- precision: 0.969
- recall: 0.958
- f1: 0.963
- roc_auc: 0.989
df_biases_pruned = load_biases(file=os.path.join(temp_weights, "pruned_weights"), folds=5)
df_biases_pruned.sort_values(by="abs_bias", ascending=False).head(10)
| bias | abs_bias | pow2_bias | ||
|---|---|---|---|---|
| fold | gene | |||
| 4 | RUNX1 | -2.475160 | 2.475160 | 6.126415 |
| SUZ12.EZH2 | -2.461908 | 2.461908 | 6.060989 | |
| 2 | SUZ12.EZH2 | -2.451049 | 2.451049 | 6.007642 |
| 1 | SUZ12.EZH2 | -2.388376 | 2.388376 | 5.704338 |
| 3 | PRKCD | -2.239764 | 2.239764 | 5.016545 |
| RUNX1 | 2.021947 | 2.021947 | 4.088270 | |
| PRC2 | -1.983842 | 1.983842 | 3.935630 | |
| 4 | TAL1 | -1.962536 | 1.962536 | 3.851547 |
| 0 | ZAP70 | 1.947543 | 1.947543 | 3.792922 |
| TCR | 1.871278 | 1.871278 | 3.501682 |
df_biases_prunedr = df_biases_pruned.copy().reset_index()
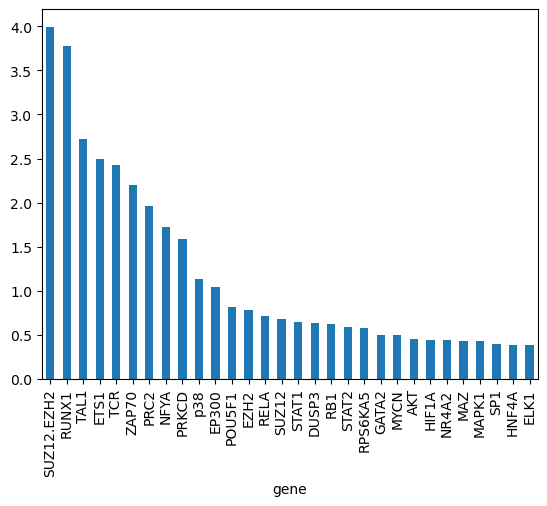
gene_biases_score = df_biases_prunedr.groupby("gene")["pow2_bias"].mean().sort_values(ascending=False)
gene_biases_score
gene
SUZ12.EZH2 3.994649e+00
RUNX1 3.776556e+00
TAL1 2.716972e+00
ETS1 2.500564e+00
TCR 2.426713e+00
...
KMT2D 5.081624e-03
TBP 1.791134e-03
HMT 4.137235e-04
ASH2L 3.378770e-06
GATA3 7.418886e-09
Name: pow2_bias, Length: 170, dtype: float32
gene_biases_score.head(30).plot.bar()
<Axes: xlabel='gene'>
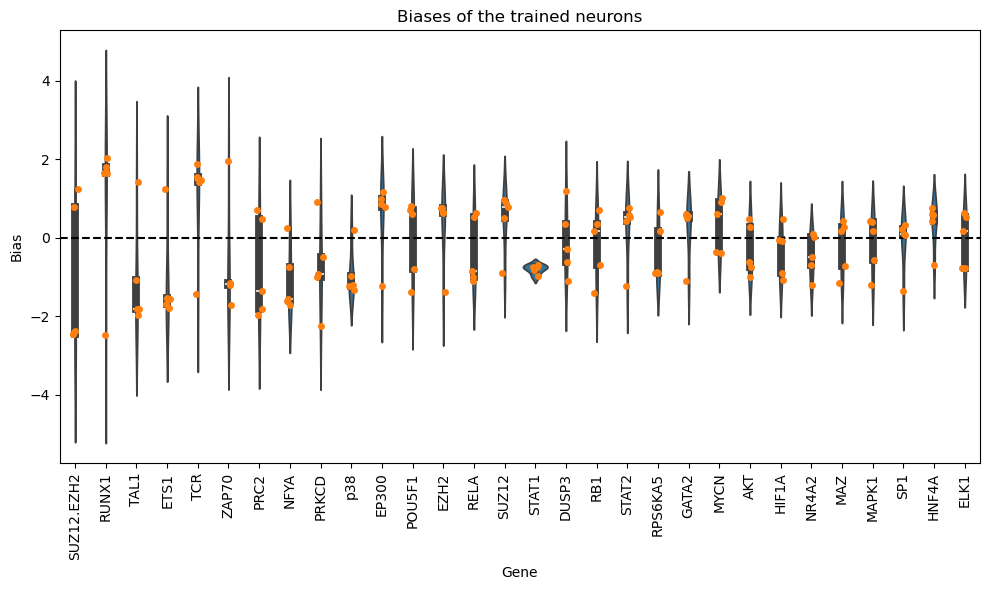
# Get the top genes sorted by mean bias
top_genes = gene_biases_score.head(30).index
# Filter the DataFrame to include only rows with these top genes
filtered_df = df_biases_prunedr[df_biases_prunedr["gene"].isin(top_genes)]
plt.figure(figsize=(10, 6))
sns.violinplot(data=filtered_df, x="gene", y="bias", order=top_genes, dodge=True)
sns.stripplot(data=filtered_df, x="gene", y="bias", order=top_genes, dodge=True)
plt.xticks(rotation=90)
plt.title("Biases of the trained neurons")
plt.xlabel("Gene")
plt.ylabel("Bias")
plt.axhline(0, linestyle="--", color="k")
plt.tight_layout()
param_compression = (1 - (pruned_models[0].count_params() / models[0].count_params())) * 100
print(f"Parameter compression: {param_compression:.2f}%")
Parameter compression: 51.22%
perf_degradation = (
(np.mean(metrics["roc_auc"]) - np.mean(pruned_metrics["roc_auc"])) / np.mean(metrics["roc_auc"])
) * 100
print(
f"Degradation in ROC-AUC after compression (positive = decrease in performance, negative = increase in performance): {perf_degradation:.2f}%"
)
Degradation in ROC-AUC after compression (positive = decrease in performance, negative = increase in performance): -0.19%